Research Projects
Isoform Admixture Model
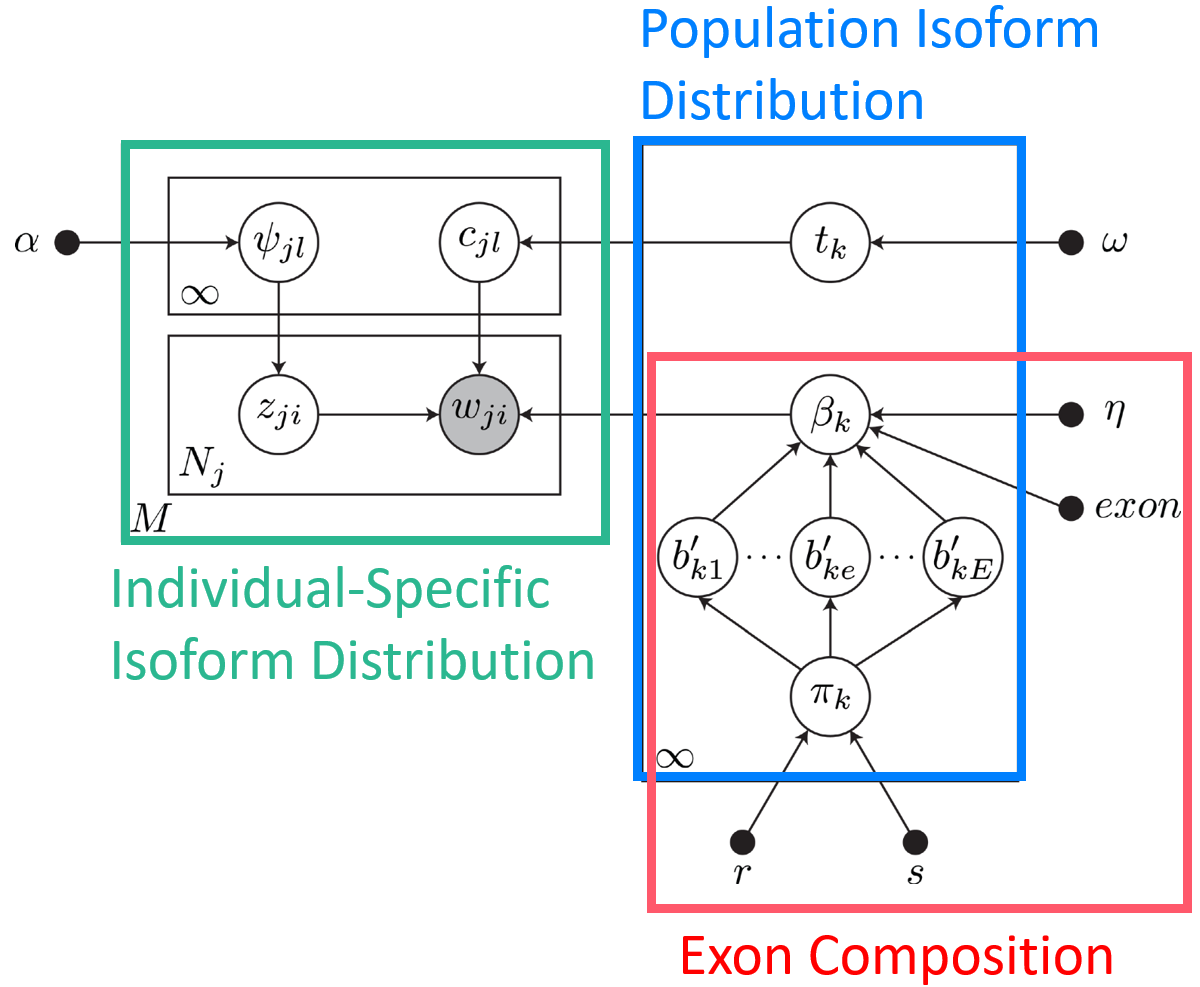
Most human protein-coding genes can be transcribed into multiple distinct mRNA isoforms. These alternative splicing patterns encourage molecular diversity, and dysregulation of isoform expression plays an important role in disease. But, isoforms are difficult to characterize from short-read RNA-seq data because they share identical subsequences and occur in different frequencies across tissues and samples. We developed BIISQ, a Bayesian nonparametric model for Isoform discovery and Individual Specific Quantification from short-read RNA-seq data.
Highlights
- [manuscript] Bayesian nonparametric discovery of isoforms and individual specific quantification
- Software
Haplotype Cluster Graphs
Genetic models cluster genetic sequences by allelic similarity as a proxy for ancestry. These models form the base for population genetics problems, for example, infer demography, resolve haplotype phase, impute missing genotypes, or identify selection. We developed the haplotype cluster graph genetic model that computes graphs where clusters of haplotypes are separated by ancestral recombination and mutations. Updates on the code and manuscript coming soon.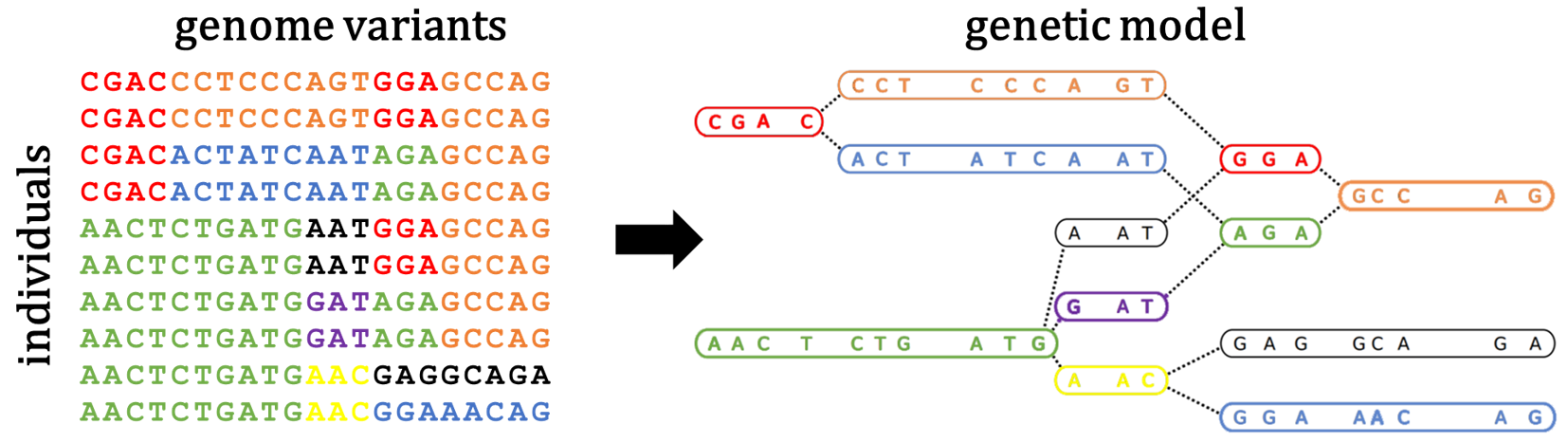
Haplotype Assembly: HapCompass
The genome sequence of a human individual can be modeled as 23 pairs of sequences of four nucleotide bases, A, C, G and T. However, ~99.5% of any two individuals’ genome sequences is shared within a population. The remaining ~0.5% of the nucleotide bases vary within a population. The sequences of these genomic variants with the non-varying DNA removed is referred to as a haplotype. Reference-based genome assembly algorithms produce assemblies where the DNA of homologous chromosomes is mixed. But, sequence reads are derived from a single haploid fragment and thus provide valuable phase information when they contain two or more variants. The haplotype assembly problem aims to compute the haplotype sequences for each chromosome given a set of aligned sequence reads to the genome and variant information. The haplotype phase of variants is inferred from assembling overlapping sequence reads.
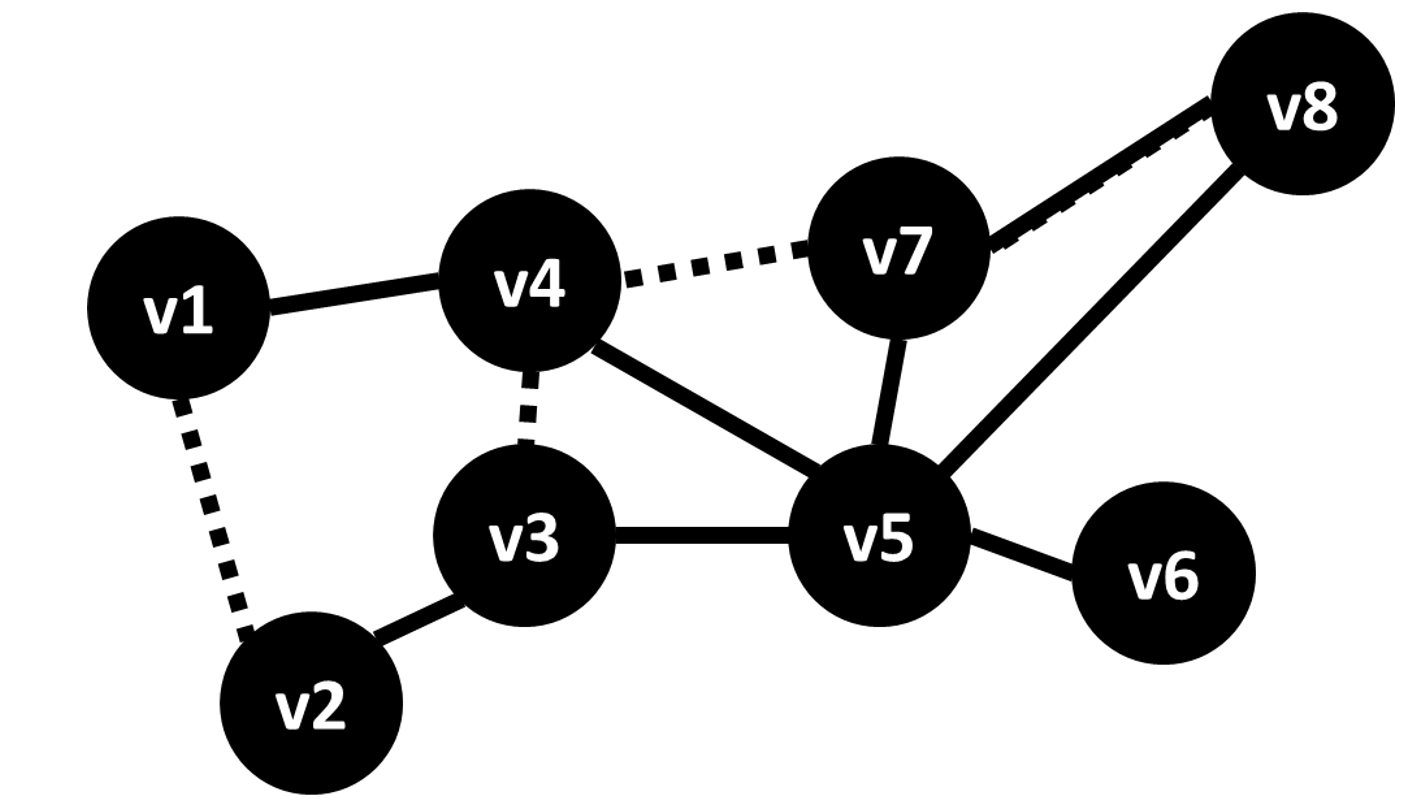
Our group developed HapCompass an algorithm for haplotype assembly that operates on a graph where variants are nodes and edges are defined by sequence reads and viewed as supporting evidence of co-occuring SNP alleles in a haplotype. Haplotype phasings correspond to spanning trees and each spanning tree uniquely defines a cycle basis. We define new optimization problems, analysis of their theoretical complexity, and a formalization of the polyploid haplotype assembly problem.
Highlights
- [manuscript] HapCompass: A Fast Cycle Basis Algorithm for Accurate Haplotype Assembly of Sequence Data
- [manuscript] Haplotype assembly in polyploid genomes and identical by descent shared tracts
- Software
Identity-by-descent: Tractatus
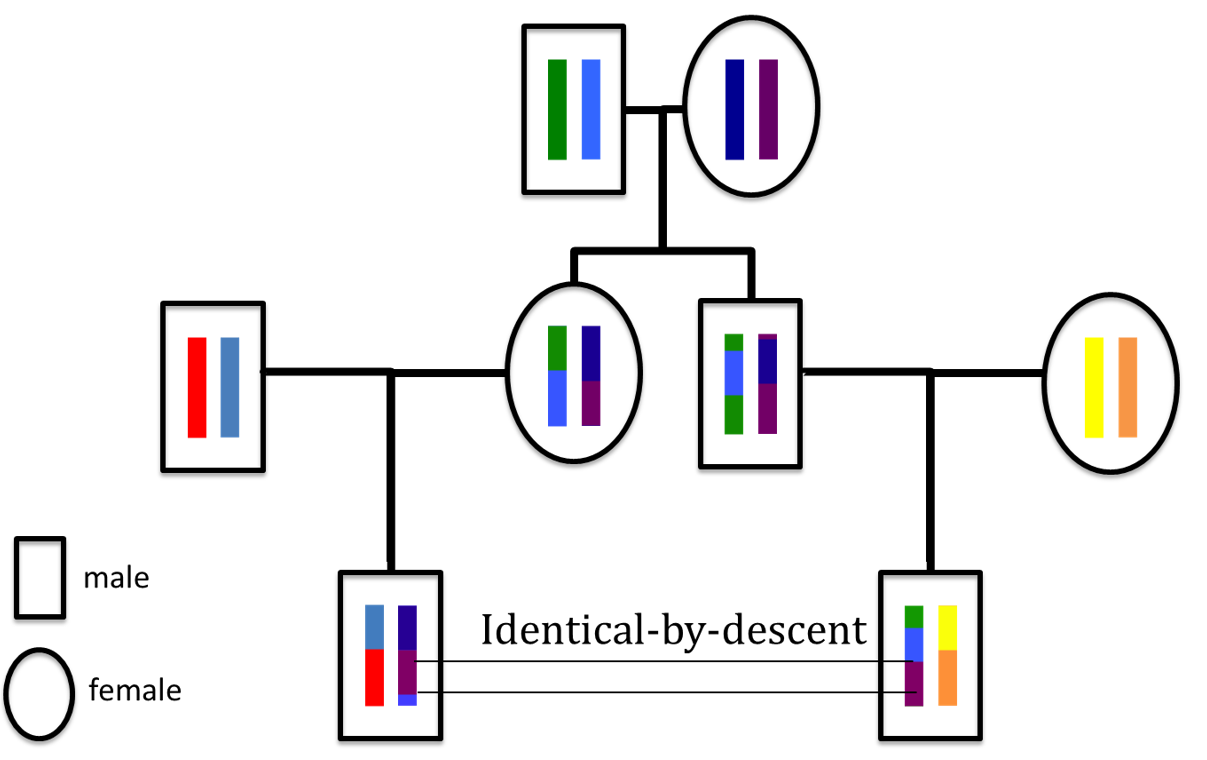
Characterizing patters of linkage disequilibrium in a genome-wide population sample is a major challenge in population genomics. It is an important first step to imputing missing data, characterizing relatedness, and inferring recombination or selection. We developed Tractatus, an exact algorithm for computing all IBD haplotype tracts in time linear in the size of the input. Tractatus resolves a long standing open problem, breaking optimally the (worst-case) quadratic time barrier of previous methods often cited as a bottleneck in haplotype analysis of genome-wide association study-sized data.
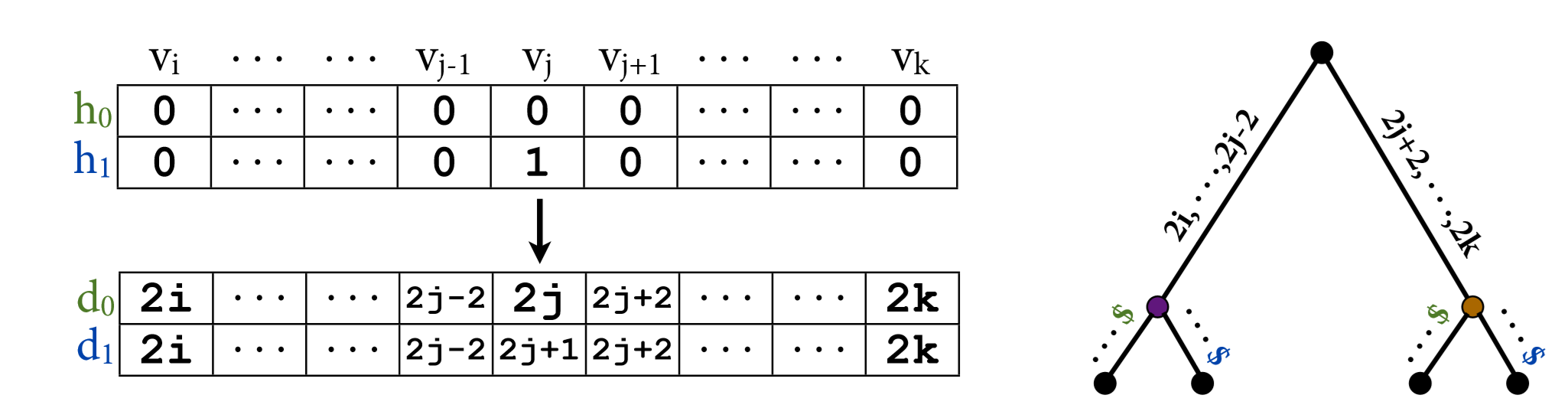
Highlights
- [author manuscript] Tractatus: an exact and subquadratic algorithm for inferring identity-by-descent multi-shared haplotype tracts
- Software
Variant Calling

Our work on variant calling was focused on computing deletions in genotype array data with applications to Autism. We developed a method that implements exact algorithms for inferring regions of hemizygosity containing genomic deletions of all sizes and frequencies in genotype data based on Mendelian inheritance patterns.
Highlights
- [manuscript] DELISHUS: an efficient and exact algorithm for genome-wide detection of deletion polymorphism in autism
- Software
Haplotype Phasing

Genome-wide association studies identify a number of individuals carrying a disease or a trait and comparing these individuals to those that do not or are not known to carry the disease/trait. Both sets of individuals are then genotyped for a large number of genetic variants which are then tested for association to the disease/trait. Current technologies, suitable for large-scale variant screening only yield the genotype information at each SNP site. The actual haplotypes in the typed region can only be obtained at a considerably high experimental cost or via haplotype phasing.
Highlights
Miscellaneous
Our group actively collaborates with professionals in many disciplines to solve problems in many areas outside of computer science, including life sciences, law, and sociology.ML and the Law
Our group is working on several problems at the intersection of Machine Learning and law. We are particularly interested in modelling court cases and interpretability in legal settings in Connecticut.
Other Work
- [manuscript] Quantitative reference transcriptomes for Nematostella vectensis early embryonic development and Crassostrea virginica in response to bacterial challenge
- [contact for details] Immunogenomics - alternative interpretations of the genetic code
- [contact for details] Exact algorithms to optimize probe selection for HIV allele-specific PCR